
Nursing Management in Common Childhood Diseases
Respiratory System: Identification and Nursing Management of Congenital Malformations
Introduction to Congenital Respiratory Malformations
Congenital respiratory malformations are rare but significant developmental anomalies that occur during fetal lung development. These conditions affect approximately 1 in 2,000-5,000 live births and can range from mild, asymptomatic cases to severe, life-threatening conditions requiring immediate intervention. As a nurse caring for children with these conditions, understanding the pathophysiology, clinical presentation, and management principles is crucial for providing high-quality care.
Key Points
- Congenital respiratory malformations result from abnormal lung development during various embryological stages
- Early diagnosis and appropriate management are crucial to prevent complications
- Nursing care focuses on respiratory support, monitoring for complications, and family education
- Most common conditions include Congenital Pulmonary Airway Malformation (CPAM), Congenital Diaphragmatic Hernia (CDH), and Congenital Lobar Emphysema (CLE)
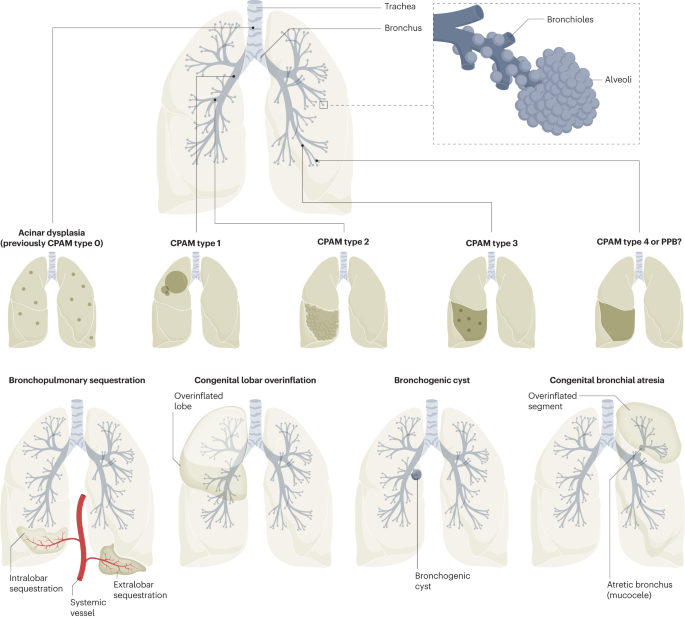
Figure 1: Overview of congenital respiratory malformations and their embryological origins
| Congenital Malformation | Incidence | Key Features | Primary Nursing Concerns |
|---|---|---|---|
| Congenital Pulmonary Airway Malformation (CPAM) | 1 in 10,000-35,000 births | Cystic masses in lung tissue with abnormal bronchial communication | Respiratory distress, infection prevention, post-surgical care |
| Congenital Diaphragmatic Hernia (CDH) | 1 in 2,500-4,000 births | Defect in diaphragm allowing abdominal contents into thoracic cavity | Severe respiratory compromise, pulmonary hypertension, nutritional support |
| Congenital Lobar Emphysema (CLE) | 1 in 20,000-30,000 births | Hyperinflation of one or more lung lobes due to air trapping | Progressive respiratory distress, post-lobectomy care, monitoring for respiratory complications |
Congenital Pulmonary Airway Malformation (CPAM)
Pathophysiology
CPAM (formerly known as Congenital Cystic Adenomatoid Malformation) is characterized by an overgrowth of terminal bronchioles forming cystic masses. During embryonic development, abnormal signaling leads to dysregulated growth of the lung tissue. The affected areas have abnormal connection to the bronchial tree and often have poor blood supply.
According to the Stocker classification, CPAM has five subtypes:
| Type | Frequency | Origin | Characteristics | Prognosis |
|---|---|---|---|---|
| Type 0 | Rare | Trachea/Bronchi | Bronchial-type airways with cartilage, smooth muscles, and glands | Incompatible with life |
| Type 1 | 70% of cases | Bronchi | Large multiloculated cysts (>2cm), lined with pseudostratified columnar epithelium | Excellent following resection, rare malignant potential |
| Type 2 | Second most common | Bronchiolar regions | Multiple smaller cysts (<2cm), often associated with other anomalies | Depends on associated anomalies, no malignant potential |
| Type 3 | Less common | Bronchiolar regions | Solid appearance with microscopic cysts, involves entire lobe | Good, no malignant potential, may have pulmonary hypertension |
| Type 4 | Very rare | Acinar structures | Peripheral thin-walled cysts, lined by alveolar type 1 or 2 cells | Strong association with pleuropulmonary blastoma |

Figure 2: Different types of Congenital Pulmonary Airway Malformation (CPAM)
Clinical Presentation
The presentation of CPAM varies widely depending on the size, location, and type of the lesion:
- Prenatal diagnosis: Increasingly common due to routine ultrasonography
- Neonatal period: Respiratory distress, tachypnea, cyanosis, retractions
- Infancy: Recurrent respiratory infections, wheezing, feeding difficulties
- Later childhood: May remain asymptomatic until infection develops
Warning Signs
Be alert for the following signs that may indicate deterioration in a child with CPAM:
- Sudden increase in respiratory rate or work of breathing
- Decreased oxygen saturation despite supplemental oxygen
- Asymmetrical chest movements
- Development of fever or increased secretions (may indicate infection in the malformation)
- Hemoptysis (rare but serious)
Diagnosis
- Prenatal: Routine ultrasonography (20-week scan), fetal MRI in complex cases
- Postnatal: Chest X-ray, CT scan with contrast (gold standard), sometimes MRI
- Histopathology: Definitive diagnosis following surgical resection

Figure 3: AP view of chest radiograph showing left lower lobe CPAM
Nursing Assessment
BREATHE Assessment for CPAM
Use this mnemonic to guide your nursing assessment of children with CPAM:
- B – Breathing pattern and rate
- R – Retractions and respiratory effort
- E – Expansion of chest (symmetry)
- A – Auscultation (breath sounds)
- T – Tachycardia (assess heart rate)
- H – Hypoxemia (oxygen saturation)
- E – Engagement with feeding and activity
Conduct a thorough respiratory assessment, including:
- Respiratory rate, pattern, and work of breathing
- Pre- and post-ductal oxygen saturations
- Breath sounds (note areas of decreased breath sounds or abnormal sounds)
- Signs of respiratory distress: nasal flaring, grunting, retractions
- Chest wall movement and symmetry
- Cyanosis (central or peripheral)
- Feeding tolerance and patterns
- Growth parameters
Nursing Management
Nursing management of CPAM focuses on respiratory support, preventing complications, and preparing for surgical intervention when indicated.
CYSTS Mnemonic for CPAM Nursing Care
- C – Careful respiratory monitoring and support
- Y – Yield optimal nutritional support
- S – Surgical preparation and post-operative care
- T – Therapeutic positioning
- S – Support family and provide education
Respiratory Support
- Monitor oxygen saturation continuously in symptomatic infants
- Administer oxygen as prescribed to maintain saturation >92% (or as ordered)
- Position with affected side elevated to optimize ventilation
- Prepare for and assist with intubation if severe respiratory distress
- Monitor ventilator settings and ensure proper function
- Perform chest physiotherapy as ordered
- Suction airway as needed to maintain patency
Preventing Infection
- Monitor for signs of infection (fever, increased respiratory distress, purulent secretions)
- Administer prophylactic antibiotics as prescribed
- Maintain aseptic technique during procedures
- Educate family about infection prevention measures
Surgical Management
- Prepare infant for surgery (lobectomy most common)
- Provide pre-operative care: NPO status, IV access, lab work
- Manage post-operative care:
- Monitor vital signs, oxygen saturation, and respiratory status
- Assess chest tubes for proper functioning and drainage
- Provide pain management as prescribed
- Monitor surgical site for bleeding, infection
- Resume feeding as tolerated
Family Education and Support
- Explain the condition, surgical procedure, and expected outcomes
- Teach recognition of respiratory distress signs
- Provide anticipatory guidance for post-discharge care
- Connect family with support resources
- Educate on importance of follow-up appointments
Long-term Monitoring and Outcomes
Children with CPAM require long-term follow-up to monitor lung function and development:
- Regular pulmonary function tests for children old enough to cooperate
- Chest X-rays to monitor remaining lung tissue
- Monitoring for recurrent respiratory infections
- Growth and development assessments
- Prognosis is generally excellent after surgical resection
- Type 1 and Type 4 lesions may require long-term monitoring due to malignant potential
Congenital Diaphragmatic Hernia (CDH)
Pathophysiology
Congenital diaphragmatic hernia (CDH) occurs when the diaphragm fails to close properly during embryonic development, creating an opening that allows abdominal organs to herniate into the chest cavity. This herniation compresses the developing lungs, leading to pulmonary hypoplasia (underdeveloped lungs) and often pulmonary hypertension.
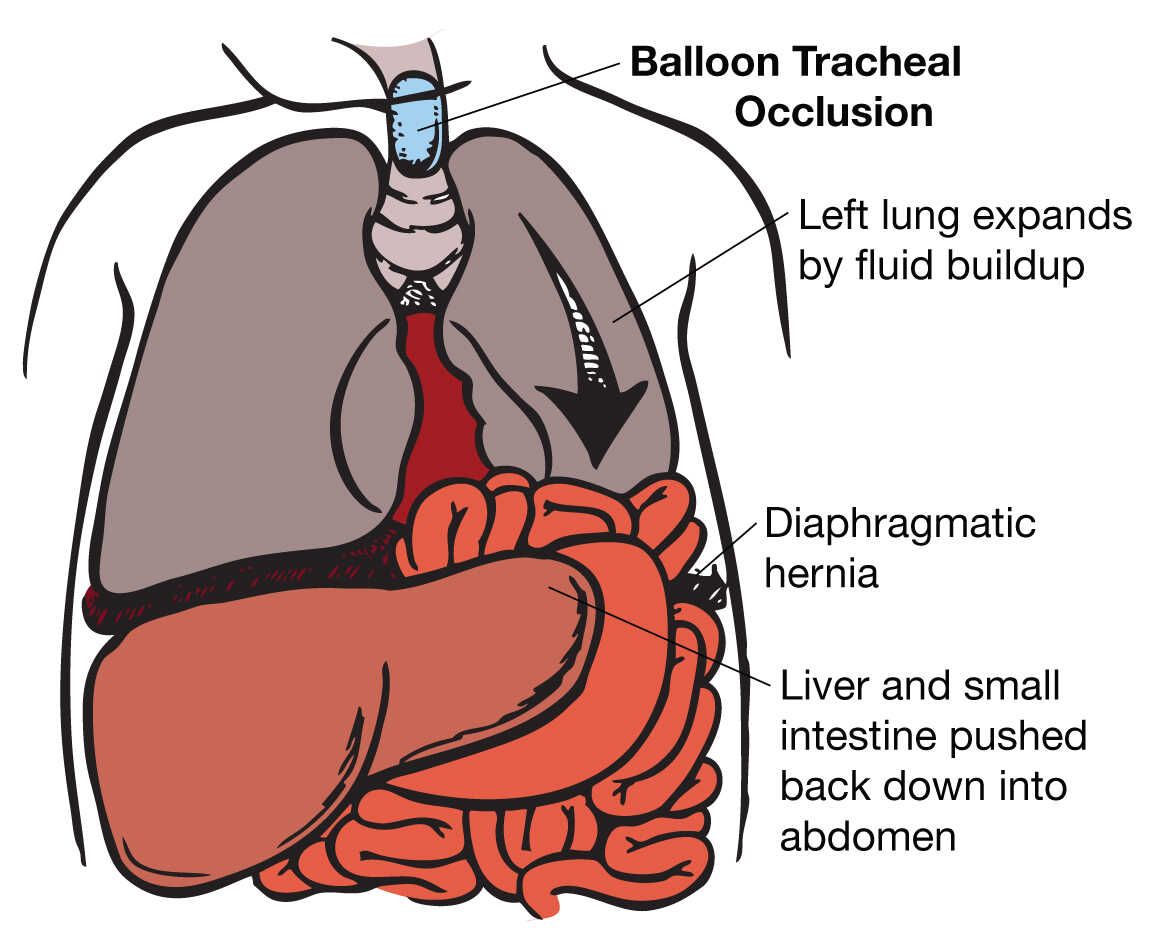
Figure 4: Illustration of Congenital Diaphragmatic Hernia showing abdominal contents in the thoracic cavity
There are three main types of CDH:
- Bochdalek hernia (posterolateral): Most common (80-90%), usually occurs on the left side
- Morgagni hernia (retrosternal): Less common (2-3%), usually right-sided
- Central tendon defect: Rare
The severity of CDH depends on:
- Size and location of the defect
- Timing of herniation during fetal development
- Organs involved (stomach, intestines, liver, spleen)
- Degree of pulmonary hypoplasia
- Presence of associated anomalies (cardiac, CNS, chromosomal)
Clinical Presentation
CDH typically presents in the immediate newborn period with:
- Respiratory distress: Severe, immediate onset after birth
- Scaphoid abdomen: Flattened or concave due to abdominal contents in thorax
- Asymmetric chest expansion: Reduced on affected side
- Displaced heart sounds: Shifted away from affected side
- Bowel sounds in chest: May be auscultated over thorax
- Cyanosis: Often refractory to oxygen
- Decreased breath sounds on affected side
Critical Alert
CDH is a medical emergency. Bag-mask ventilation should be avoided as it may cause gastric distention, further compressing the lungs. Immediate endotracheal intubation with gentle ventilation is preferred.
Diagnosis
- Prenatal: Routine ultrasound may detect the defect, with follow-up MRI to assess severity
- Postnatal: Chest X-ray showing intestinal loops in thorax, shifted mediastinum
- Additional studies: Echocardiography to assess cardiac function and rule out associated cardiac defects
Nursing Assessment
DIAPHRAGM Assessment for CDH
Use this mnemonic to guide your nursing assessment of infants with CDH:
- D – Distress (respiratory) assessment
- I – Intubation status and ventilator settings
- A – Abdomen (scaphoid appearance)
- P – Pulmonary hypertension signs
- H – Heart position and sounds
- R – Respiratory effort and breath sounds
- A – Arterial blood gases
- G – Gastric decompression (OG/NG tube function)
- M – Monitoring pre- and post-ductal saturations
Perform a comprehensive assessment, focusing on:
- Respiratory status: rate, effort, breath sounds, chest movement
- Pre- and post-ductal oxygen saturations (to assess right-to-left shunting)
- Heart rate, blood pressure, perfusion
- Abdominal appearance and bowel sounds
- Presence and function of tubes: endotracheal, orogastric/nasogastric, chest tubes
- Pain and comfort level
- Laboratory values: blood gases, electrolytes, complete blood count
Nursing Management
Nursing management of CDH is complex and focuses on stabilization before surgical repair.
HERNIA Mnemonic for CDH Nursing Care
- H – Hypoxemia management and oxygenation
- E – Endotracheal tube and ventilator management
- R – Reduction of pulmonary hypertension
- N – Nasogastric/orogastric tube decompression
- I – Intravenous access and fluid management
- A – Assessment and monitoring of vital signs
Initial Stabilization
- Assist with immediate endotracheal intubation following delivery
- Place orogastric tube to low continuous suction to decompress stomach
- Monitor pre- and post-ductal oxygen saturations
- Begin oxygen therapy (typically starting at 50%, adjusting based on saturations)
- Goal saturations: >65% at 5 minutes, >75% at 10 minutes of life
- Avoid bag-mask ventilation to prevent further lung compression
Respiratory Management
- Manage ventilator settings using “gentle ventilation” strategy to avoid barotrauma
- Typical ventilator settings:
- SIMV rate 30-40
- PIP <25 cmH2O
- Inspiratory time 0.35 seconds
- Tidal volume <5cc/kg
- Start oxygen at 50%, may wean at 6 hours if stable with preductal sats >85%
- Consider high-frequency oscillatory ventilation (HFOV) if conventional ventilation is insufficient
- Monitor end-tidal CO2 and transcutaneous monitors
- Prepare for possible inhaled nitric oxide (iNO) therapy for pulmonary hypertension
- Maintain endotracheal tube position and security
- Perform airway suctioning as needed with caution
Hemodynamic Management
- Target mean arterial pressure of 40-50 mmHg in term infants
- Administer fluid resuscitation judiciously (restricted to <80 mL/kg/day)
- Assist with placement of umbilical or peripheral arterial lines for BP monitoring
- Administer vasopressors as prescribed (often dopamine as first-line)
- Monitor for signs of pulmonary hypertension and prepare for treatment
- Maintain adequate sedation to minimize respiratory effort and oxygen consumption
Surgical Preparation and Postoperative Care
- Prepare for surgery once physiologically stable (may be days to weeks)
- Preoperative preparation:
- Maintain NPO status
- Ensure laboratory tests are completed (CBC, electrolytes, type and cross)
- Administer preoperative antibiotics as ordered
- Postoperative care:
- Monitor for signs of compartment syndrome (decreased distal pulses, abdominal distention, decreased urine output)
- Maintain gastric decompression with OG/NG tube
- Monitor chest tubes if present
- Provide pain management
- Monitor for respiratory deterioration
- Gradual introduction of enteral feeds when bowel function returns
ECMO Considerations
Extracorporeal Membrane Oxygenation (ECMO) may be required for infants with severe CDH who fail conventional management. Nursing considerations include:
- Recognize indications for ECMO:
- Inability to maintain preductal saturations >85%
- Severe respiratory acidosis (pH <7.15)
- High ventilator requirements (PIP >28 cmH2O, MAP >15 cmH2O)
- Evidence of inadequate oxygen delivery (lactate >5)
- Refractory hypotension
- Prepare for ECMO cannulation
- Monitor ECMO circuit and parameters
- Provide specialized care for the ECMO patient
Family Support and Education
- Explain the condition, interventions, and expected course
- Provide emotional support through this critical illness
- Encourage parental bonding despite the critical care environment
- Facilitate kangaroo care when appropriate
- Educate about long-term follow-up needs
- Connect with support resources for families of children with CDH
Long-term Monitoring and Outcomes
After discharge, children with CDH require comprehensive follow-up:
- Regular growth and development monitoring
- Pulmonary function tests
- Cardiac evaluation for persistent pulmonary hypertension
- Monitoring for gastroesophageal reflux (common in CDH)
- Nutritional support for feeding difficulties
- Assessment for scoliosis and chest wall deformities
- Neurodevelopmental evaluation
- Hearing assessment (especially if ECMO was required)
Prognosis depends on severity of pulmonary hypoplasia, associated anomalies, and complications. Survival rates have improved to 70-90% with advances in care.
Congenital Lobar Emphysema (CLE)
Pathophysiology
Congenital lobar emphysema (CLE), also known as congenital lobar overinflation, is characterized by hyperinflation of one or more lung lobes without destruction of alveolar walls. This occurs due to a “ball-valve” mechanism that allows air to enter during inspiration but prevents complete expiration, leading to progressive air trapping.
The primary causes include:
- Bronchial cartilage deficiency or malformation (most common, 25% of cases)
- Bronchial obstruction (internal or external)
- Alveolar abnormalities (polyalveolar lobe)
- Idiopathic (approximately 50% of cases)
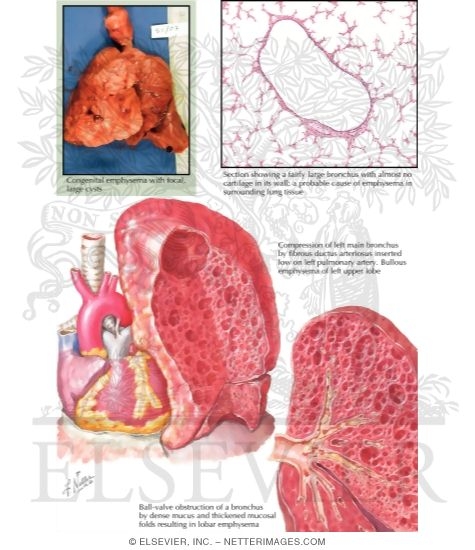
Figure 5: Illustration of Congenital Lobar Emphysema showing hyperinflated lobe compressing adjacent lung tissue
The most commonly affected lobes are:
- Left upper lobe (43%)
- Right middle lobe (32%)
- Right upper lobe (21%)
- Lower lobes (rarely affected, ~2%)
Clinical Presentation
CLE can present with varying severity:
- Neonatal period (33% symptomatic at birth): Severe respiratory distress, cyanosis
- Early infancy (most diagnosed by 6 months): Progressive respiratory distress, recurrent infections
- Older children/adults: May be asymptomatic or have subtle symptoms
Common clinical findings include:
- Tachypnea and respiratory distress
- Diminished breath sounds over affected area
- Hyperresonance on percussion of affected side
- Wheezing or rhonchi
- Asymmetric chest expansion
- Feeding difficulties
- Failure to thrive
- Cyanosis (in severe cases)
Associated conditions (in 14-20% of cases):
- Congenital heart disease (ventricular septal defect, patent ductus arteriosus)
- Renal anomalies
- Pectus excavatum
Warning Signs
Monitor for these signs of worsening CLE:
- Rapidly increasing respiratory rate or work of breathing
- Progressive cyanosis or declining oxygen saturations
- Increased chest asymmetry
- Mediastinal shift (may be detected on examination or imaging)
- Hemodynamic instability (compensatory tachycardia, hypotension)
Diagnosis
- Chest X-ray: Shows hyperinflated, hyperlucent lobe, mediastinal shift, compressed adjacent lung
- CT scan: Gold standard for diagnosis, shows extent of hyperinflation and compression
- Bronchoscopy: May be performed to rule out foreign body or other obstructive lesions
- Ventilation-perfusion scan: Shows decreased perfusion in affected lobe

Figure 6: Chest radiograph showing characteristic findings of congenital lobar emphysema
Nursing Assessment
LOBAR Assessment for CLE
Use this mnemonic to guide your nursing assessment of children with CLE:
- L – Labored breathing and respiratory effort
- O – Oxygen saturation monitoring
- B – Breath sounds (diminished over affected area)
- A – Asymmetry of chest movement
- R – Resonance (hyperresonance on percussion)
Perform a comprehensive assessment focusing on:
- Respiratory status: rate, pattern, work of breathing, retractions
- Oxygen saturation and need for supplemental oxygen
- Breath sounds and chest percussion
- Chest symmetry and expansion
- Cyanosis or color changes
- Feeding ability and tolerance
- Growth parameters and development
- Signs of respiratory infection
Nursing Management
Management of CLE depends on symptom severity:
- Asymptomatic/Mild symptoms: Conservative management with monitoring
- Moderate to Severe symptoms: Surgical intervention (lobectomy)
EMPHYSEMA Mnemonic for CLE Nursing Care
- E – Effective respiratory monitoring
- M – Medications and oxygen administration
- P – Positioning to optimize breathing
- H – Hydration and nutrition management
- Y – Yield optimal surgical outcomes (pre/post-op care)
- S – Support family coping
- E – Education about the condition and care
- M – Monitor for complications
- A – Assess developmental progress
Conservative Management
- Regular monitoring of respiratory status
- Position with affected side down to improve ventilation to unaffected lung
- Oxygen therapy as needed
- Monitor for infections and provide prompt treatment
- Regular follow-up with chest X-rays to monitor progression
- Ensure adequate nutrition and hydration
- Developmental monitoring
Surgical Management (Lobectomy)
For infants with significant symptoms, surgical removal of the affected lobe (lobectomy) is the standard treatment.
Preoperative Nursing Care:
- Prepare for surgical intervention (lobectomy)
- Ensure adequate respiratory support
- Maintain NPO status as ordered
- Obtain necessary laboratory tests
- Establish reliable IV access
- Administer preoperative medications as ordered
- Provide emotional support to family
- Document baseline assessment
Postoperative Nursing Care:
- Monitor vital signs, respiratory status, and oxygen saturation closely
- Assess chest tube function and drainage
- Provide pain management
- Monitor for surgical complications:
- Bleeding
- Pneumothorax
- Infection
- Atelectasis
- Encourage deep breathing and early mobilization
- Resume feeding as tolerated
- Monitor fluid and electrolyte balance
- Provide wound care
Family Education and Support
- Explain the condition and treatment plan
- Provide pre- and post-operative teaching
- Teach recognition of respiratory distress signs
- Provide discharge instructions:
- Pain management
- Wound care
- Activity restrictions
- Follow-up appointments
- When to seek medical attention
- Support family coping with diagnosis and treatment
- Provide resources for home care
Long-term Monitoring and Outcomes
The prognosis for CLE is generally excellent after appropriate treatment:
- Regular follow-up to monitor lung growth and function
- Pulmonary function testing in older children
- Monitoring for compensatory lung growth
- Respiratory infection prevention
- Most children have normal lung function and exercise tolerance
- Lung growth continues throughout childhood, compensating for removed lobe
Comparative Analysis of Congenital Respiratory Malformations
| Feature | CPAM | CDH | CLE |
|---|---|---|---|
| Pathophysiology | Abnormal development of terminal bronchioles with cystic masses | Defect in diaphragm allowing abdominal contents into thorax | Hyperinflation of lobe due to air trapping from bronchial abnormality |
| Incidence | 1 in 10,000-35,000 births | 1 in 2,500-4,000 births | 1 in 20,000-30,000 births |
| Most Common Location | Lower lobes | Left posterolateral (Bochdalek) | Left upper lobe |
| Primary Clinical Presentation | Variable: asymptomatic to respiratory distress | Severe respiratory distress at birth | Progressive respiratory distress in early infancy |
| Key Diagnostic Finding | Cystic or solid masses on imaging | Abdominal organs in thoracic cavity | Hyperinflated, hyperlucent lobe with mediastinal shift |
| Primary Treatment | Surgical resection of affected area | Surgical repair of diaphragmatic defect | Lobectomy for symptomatic cases |
| Critical Nursing Care | Respiratory support, infection prevention | Ventilation management, pulmonary hypertension treatment | Respiratory monitoring, positioning |
| Prognosis | Excellent after resection | Variable, depends on pulmonary hypoplasia severity | Excellent after lobectomy |
| Long-term Concerns | Malignant potential (Type 1 and 4) | Pulmonary hypertension, feeding issues, developmental delays | Generally normal lung function |
Summary of Nursing Priorities
Nursing Priorities Across All Congenital Respiratory Malformations
- Maintain adequate oxygenation and ventilation
- Consistently assess respiratory status
- Position to optimize respiratory function
- Administer oxygen and respiratory support as needed
- Monitor for and prevent complications
- Recognize early signs of respiratory deterioration
- Implement infection prevention measures
- Monitor for condition-specific complications
- Provide appropriate pre- and post-operative care
- Prepare patients and families for surgical interventions
- Deliver evidence-based post-operative care
- Manage pain effectively
- Support optimal nutrition and growth
- Coordinate feeding strategies with respiratory status
- Monitor growth parameters
- Adapt feeding approaches based on condition
- Provide comprehensive family education and support
- Educate about the condition, treatment, and home care
- Teach recognition of warning signs
- Connect families with appropriate resources
- Support family involvement in care
Conclusion
Congenital respiratory malformations present unique challenges in pediatric nursing care. Understanding the pathophysiology, clinical presentation, and nursing management of these conditions enables nurses to provide optimal care for affected children. While each condition has specific management considerations, all require careful respiratory monitoring, appropriate surgical preparation and post-operative care, and comprehensive family education and support.
Early identification and intervention are key to improving outcomes. With advances in prenatal diagnosis, surgical techniques, and critical care management, the prognosis for children with these conditions has improved significantly. The nurse plays a crucial role in the interprofessional team caring for these children, from initial diagnosis through long-term follow-up.
Remember the “BREATHE” Approach for All Congenital Respiratory Malformations
- B – Be vigilant with respiratory assessment
- R – Recognize early warning signs
- E – Educate families thoroughly
- A – Adapt care to individual patient needs
- T – Treat pain and discomfort effectively
- H – Help families navigate the healthcare journey
- E – Ensure comprehensive follow-up care