
Pediatric Nursing Notes
Cardiovascular Disease, Anemia, and Leukemia
Pediatric Cardiovascular Disease: Congenital Heart Disease
Overview
Congenital heart disease (CHD) refers to structural or functional heart abnormalities present at birth that affect normal cardiac function. It is the most common type of birth defect, affecting approximately 8-10 per 1,000 live births worldwide.
Clinical Pearl
CHD is the leading cause of infant mortality related to birth defects. Early recognition and intervention significantly improve outcomes.
Classification of Congenital Heart Defects

Figure 1: Common types of congenital heart defects and the structures affected
Congenital heart defects are commonly classified into three main categories:
| Category | Description | Examples |
|---|---|---|
| Cyanotic Defects | Result in decreased pulmonary blood flow or mixing of oxygenated and deoxygenated blood, causing cyanosis | Tetralogy of Fallot, Transposition of Great Arteries, Tricuspid Atresia, Total Anomalous Pulmonary Venous Return, Truncus Arteriosus |
| Acyanotic Defects | Left-to-right shunts that increase pulmonary blood flow but don’t typically cause cyanosis | Atrial Septal Defect (ASD), Ventricular Septal Defect (VSD), Patent Ductus Arteriosus (PDA), Atrioventricular Canal Defect |
| Obstructive Defects | Obstruct blood flow through the heart or great vessels | Coarctation of Aorta, Aortic Stenosis, Pulmonary Stenosis, Hypoplastic Left Heart Syndrome |
Mnemonic: “The 5 T’s” of Cyanotic Heart Defects
T Tetralogy of Fallot
T Transposition of Great Arteries
T Truncus Arteriosus
T Tricuspid Atresia
T Total Anomalous Pulmonary Venous Return
Common Congenital Heart Defects
Tetralogy of Fallot (TOF)
Pathophysiology: TOF consists of four defects:
- Ventricular septal defect (VSD)
- Pulmonary stenosis
- Right ventricular hypertrophy
- Overriding aorta
Clinical Manifestations:
- Cyanosis (blue discoloration of skin, especially during crying or feeding)
- Hypercyanotic “tet spells” – acute episodes of worsening cyanosis, hypoxemia, and dyspnea
- Clubbing of fingers and toes
- Growth retardation
- Squatting position (often assumes this position to relieve symptoms)
Management: Surgical repair is necessary, typically performed between 3-6 months of age. For hypercyanotic “tet spells,” knee-chest position, oxygen, morphine, and propranolol may be administered.
Ventricular Septal Defect (VSD)
Pathophysiology: Abnormal opening in the septum between the heart’s ventricles, allowing blood to flow from the left to the right ventricle.
Clinical Manifestations:
- Harsh holosystolic murmur, loudest at the left sternal border
- Increased pulmonary vascularity on chest X-ray
- Tachypnea, dyspnea, and poor feeding in large defects
- Heart failure in large defects
Management: Small VSDs may close spontaneously. Larger defects may require medical management of heart failure and/or surgical repair.
Atrial Septal Defect (ASD)
Pathophysiology: Opening in the atrial septum allowing blood flow between the atria.
Clinical Manifestations:
- Often asymptomatic in children
- Fixed splitting of S2 heart sound
- Systolic ejection murmur at upper left sternal border
- Right ventricular hypertrophy on ECG
Management: Small defects may close spontaneously. Larger defects typically require closure via cardiac catheterization or surgery.
Transposition of the Great Arteries (TGA)
Pathophysiology: The aorta arises from the right ventricle, and the pulmonary artery arises from the left ventricle, creating two separate circulatory systems.
Clinical Manifestations:
- Severe cyanosis apparent shortly after birth
- Tachypnea and respiratory distress
- Absence of murmur in isolated TGA
Management: Prostaglandin E1 to maintain patent ductus arteriosus, balloon atrial septostomy to improve mixing, and definitive surgical correction (arterial switch operation) in early infancy.
Clinical Manifestations of CHD
The clinical presentation of CHD varies widely depending on the type and severity of the defect. Common signs and symptoms include:
Neonatal Presentation
- Cyanosis (bluish discoloration)
- Tachypnea (rapid breathing)
- Feeding difficulties
- Heart murmur
- Shock-like symptoms
- Absent femoral pulses (in coarctation)
Infant/Child Presentation
- Failure to thrive
- Exercise intolerance
- Recurrent respiratory infections
- Heart failure symptoms
- Clubbing of fingers
- Developmental delays
Warning Signs Requiring Immediate Attention
The following symptoms may indicate critical CHD and require immediate medical intervention:
- Sudden onset of cyanosis
- Respiratory distress
- Poor perfusion or shock-like symptoms
- Hypercyanotic spells
- Heart failure with pulmonary edema
Diagnostic Approach
| Diagnostic Test | Clinical Utility | Nursing Considerations |
|---|---|---|
| Pulse Oximetry Screening | Noninvasive test to detect hypoxemia; mandated screening for all newborns | Perform on right hand and either foot after 24 hours of life; <95% or difference >3% between extremities warrants further evaluation |
| Chest X-ray | Evaluates heart size, pulmonary vascularity, and chamber enlargement | Ensure proper positioning; may require sedation in uncooperative children |
| Electrocardiogram (ECG) | Assesses cardiac rhythm, chamber enlargement, and conduction abnormalities | Explain procedure to reduce anxiety; ensure proper lead placement |
| Echocardiography | Gold standard for diagnosis; provides detailed anatomical and functional information | Prepare child for ultrasound gel (cold sensation); may require sedation in younger children |
| Cardiac Catheterization | Provides hemodynamic information and allows for interventional procedures | Requires NPO status, IV access, monitor for complications (bleeding, infection, arrhythmias) |
| Cardiac MRI/CT | Advanced imaging for complex defects and extracardiac structures | Assess for claustrophobia; MRI requires removal of metal objects; may require sedation/anesthesia |
Management and Nursing Care
Management of CHD typically involves a multidisciplinary approach, including medical management, surgical correction, and supportive care.
Medical Management
- Prostaglandin E1: Maintains patency of ductus arteriosus in ductal-dependent lesions
- Diuretics: Manage fluid overload in heart failure
- ACE inhibitors: Reduce afterload in heart failure
- Beta-blockers: Manage arrhythmias and reduce myocardial oxygen demand
- Antibiotics: Endocarditis prophylaxis for high-risk lesions
Surgical/Interventional Approaches
- Palliative procedures: Improve symptoms and stabilize (e.g., shunt procedures)
- Definitive repair: Complete correction of defect
- Staged repair: Series of surgeries for complex defects
- Catheter-based interventions: ASD/VSD closure, balloon valvuloplasty
- Heart transplantation: For severe, uncorrectable defects
Nursing Management Tips
- Respiratory monitoring: Assess respiratory status, oxygen saturation, and work of breathing
- Cardiac monitoring: Monitor vital signs, perfusion, and early signs of cardiac decompensation
- Nutritional support: Ensure adequate caloric intake; may require high-calorie formula, frequent small feedings, or NG/G-tube feeding
- Fluid management: Monitor intake and output, daily weights, and signs of fluid overload
- Growth and development: Track growth parameters and developmental milestones
- Family education: Teach parents about the condition, medication administration, and recognition of warning signs
Mnemonic: “CARDIAC” – Key Nursing Assessments for CHD
C Color (cyanosis, pallor)
A Activity tolerance and developmental status
R Respiratory status (rate, effort, breath sounds)
D Diaphoresis with feeding or activity
I Intake and Output (feeding difficulties, fluid balance)
A Auscultation findings (murmurs, gallops, rubs)
C Capillary refill and peripheral perfusion
Pediatric Anemia
Overview
Anemia is defined as a reduction in red blood cell (RBC) mass or hemoglobin concentration below established reference values for age and sex. It is one of the most common hematological disorders in children worldwide, affecting approximately 40% of children globally.
Clinical Pearl
Anemia is not a diagnosis but a sign of an underlying condition. Always investigate the cause of anemia in a child, as it can range from nutritional deficiencies to serious hematological disorders.
Normal Hemoglobin Values in Children
| Age | Mean Hemoglobin (g/dL) | Lower Limit of Normal (g/dL) |
|---|---|---|
| Cord blood | 16.5 | 13.5 |
| 2 weeks | 16.5 | 13.0 |
| 1 month | 14.0 | 10.0 |
| 2-6 months | 12.0 | 10.0 |
| 6 months – 2 years | 12.0 | 11.0 |
| 2-6 years | 12.5 | 11.5 |
| 6-12 years | 13.5 | 11.5 |
| 12-18 years (female) | 14.0 | 12.0 |
| 12-18 years (male) | 14.5 | 13.0 |
Important Note
Physiologic anemia of infancy is a normal phenomenon occurring at 2-3 months of age, with hemoglobin dropping to 9-11 g/dL. This is not pathological and doesn’t require intervention.
Classification of Pediatric Anemia
Anemia can be classified based on red blood cell morphology (size and hemoglobin content) or etiology:
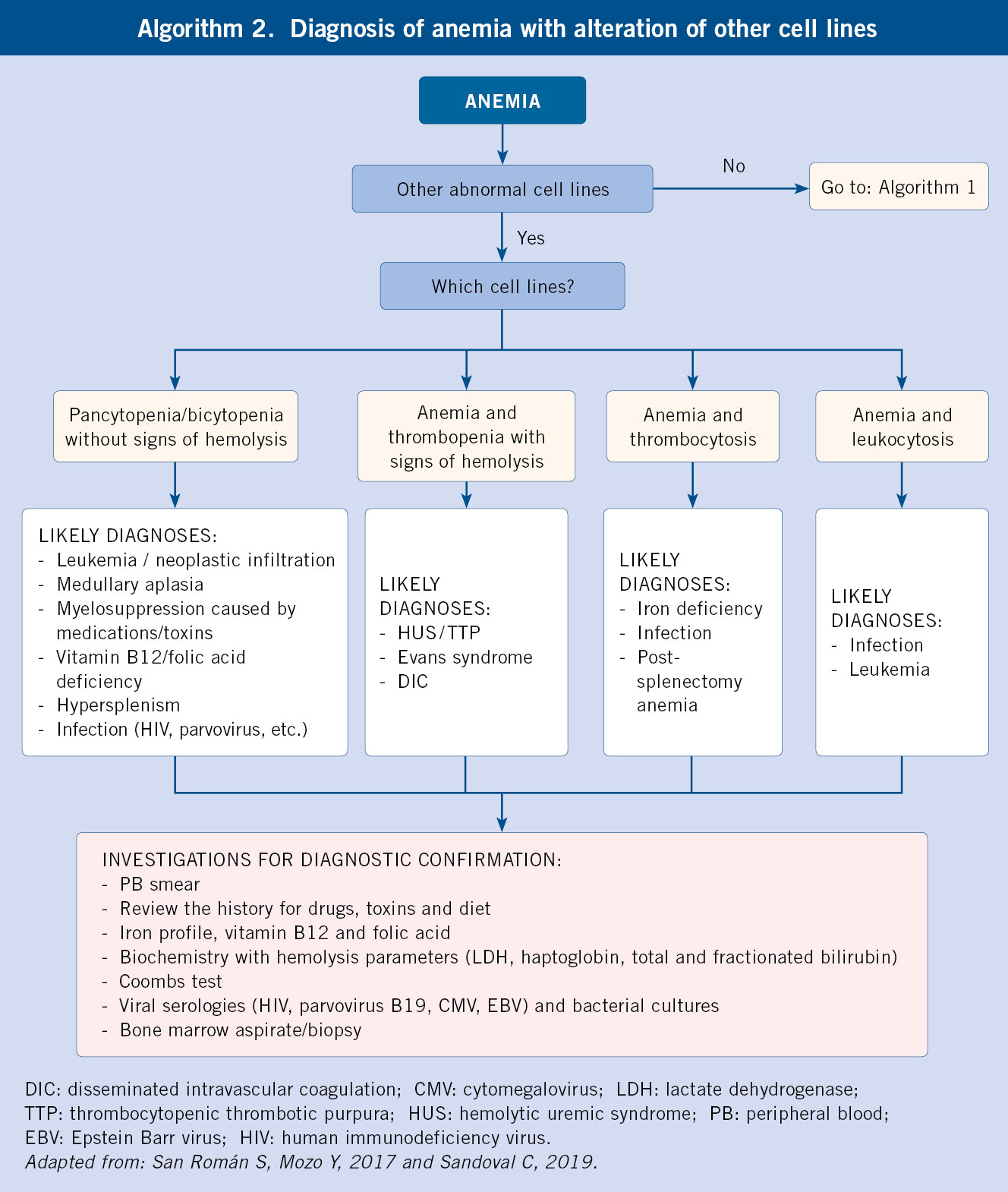
Figure 2: Diagnostic approach to pediatric anemia based on red cell indices
Microcytic Anemia (MCV ↓)
- Iron deficiency anemia
- Thalassemia
- Anemia of chronic disease
- Lead poisoning
- Sideroblastic anemia
Normocytic Anemia (Normal MCV)
- Acute blood loss
- Hemolytic anemia
- Aplastic anemia
- Chronic renal disease
- Bone marrow infiltration
Macrocytic Anemia (MCV ↑)
- Vitamin B12 deficiency
- Folate deficiency
- Liver disease
- Hypothyroidism
- Myelodysplastic syndrome
Mnemonic: “TICS” – Causes of Microcytic Anemia
T Thalassemia
I Iron deficiency
C Chronic disease/inflammation
S Sideroblastic anemia
Common Types of Pediatric Anemia
Iron Deficiency Anemia (IDA)
Pathophysiology: Inadequate iron for hemoglobin synthesis due to increased requirements, decreased intake, or blood loss.
Risk Factors:
- Rapid growth periods (infancy, adolescence)
- Exclusive breastfeeding beyond 6 months without iron supplementation
- Cow’s milk consumption before 12 months
- Poor dietary iron intake
- Chronic blood loss (GI bleeding, heavy menses)
- Prematurity or low birth weight
Clinical Manifestations:
- Pallor, fatigue, irritability
- Pica (eating non-food items like ice, dirt)
- Developmental delays
- Poor attention and school performance
- Tachycardia, systolic murmur
Laboratory Findings:
- Low hemoglobin and hematocrit
- Low MCV (microcytic)
- Low MCH (hypochromic)
- Decreased serum ferritin
- Increased TIBC (total iron-binding capacity)
- Low serum iron
Management: Oral iron supplementation (3-6 mg/kg/day of elemental iron divided 1-3 times daily), dietary counseling, and treating underlying causes.
Thalassemia
Pathophysiology: Genetic disorders causing decreased or absent synthesis of alpha or beta globin chains of hemoglobin.
Types:
- Alpha Thalassemia: Decreased or absent alpha-globin chain synthesis (4 genes)
- Beta Thalassemia: Decreased or absent beta-globin chain synthesis (2 genes)
Clinical Manifestations:
- Thalassemia Minor/Trait: Mild or no anemia, generally asymptomatic
- Thalassemia Intermedia: Moderate anemia, may require periodic transfusions
- Thalassemia Major: Severe anemia, hepatosplenomegaly, growth failure, bone deformities, iron overload
Laboratory Findings:
- Microcytic, hypochromic anemia
- Normal or elevated RBC count
- Target cells and basophilic stippling on peripheral smear
- Normal or increased serum ferritin
- Hemoglobin electrophoresis showing abnormal patterns
Management: Depends on severity—from observation (minor) to regular transfusions, iron chelation therapy, and possibly bone marrow transplantation (major).
Sickle Cell Disease (SCD)
Pathophysiology: Genetic disorder causing abnormal hemoglobin S, which polymerizes when deoxygenated, leading to sickle-shaped RBCs that can cause vaso-occlusion and hemolysis.
Clinical Manifestations:
- Vaso-occlusive pain crises
- Acute chest syndrome
- Splenic sequestration
- Aplastic crisis
- Increased susceptibility to infections
- Delayed growth and development
- Stroke and other end-organ damage
Laboratory Findings:
- Normocytic anemia
- Reticulocytosis
- Sickle cells on peripheral smear
- Positive sickle solubility test
- Hemoglobin electrophoresis showing HbS
Management: Hydroxyurea therapy, pain management, prophylactic antibiotics, pneumococcal vaccination, regular transfusions for certain complications, and bone marrow transplantation in selected cases.
Vitamin B12/Folate Deficiency
Pathophysiology: Deficiency of vitamin B12 or folate leads to impaired DNA synthesis, resulting in abnormal RBC maturation and macrocytic anemia.
Risk Factors:
- Maternal deficiency (for breastfed infants)
- Strict vegetarian/vegan diet without supplementation
- Malabsorption syndromes
- Intestinal resection
- Certain medications (methotrexate, anticonvulsants)
Clinical Manifestations:
- Pallor, fatigue
- Glossitis (smooth, red tongue)
- Poor growth
- Neurological symptoms (with B12 deficiency: paresthesias, developmental regression, irritability)
Laboratory Findings:
- Macrocytic anemia (elevated MCV)
- Hypersegmented neutrophils
- Low serum B12 or folate levels
- Elevated methylmalonic acid and homocysteine (B12 deficiency)
- Elevated homocysteine only (folate deficiency)
Management: B12 supplementation (oral or parenteral) or folate supplementation based on the deficiency, and addressing underlying causes.
Diagnostic Approach to Pediatric Anemia
A systematic approach to diagnosing anemia involves clinical assessment and laboratory testing:
Clinical Assessment
- History: Dietary intake, growth patterns, family history, ethnicity, medications, symptoms of blood loss
- Physical Exam: Pallor, jaundice, hepatosplenomegaly, lymphadenopathy, growth parameters, developmental status
Initial Laboratory Tests
- Complete Blood Count (CBC): Hemoglobin, hematocrit, RBC indices (MCV, MCH, MCHC), RBC count, reticulocyte count
- Peripheral Blood Smear: Morphology of RBCs, WBCs, and platelets
- Reticulocyte Count: Assesses bone marrow response to anemia
Additional Tests Based on Initial Findings
Microcytic Anemia
- Serum iron, TIBC, ferritin
- Hemoglobin electrophoresis
- Lead level
- Inflammatory markers (ESR, CRP)
Normocytic Anemia
- Direct Coombs test
- Hemolysis panel (LDH, haptoglobin, bilirubin)
- Renal and liver function tests
- Bone marrow examination
Macrocytic Anemia
- Serum B12 and folate levels
- Methylmalonic acid and homocysteine
- Thyroid function tests
- Liver function tests
Clinical Pearl: The Reticulocyte Count
The reticulocyte count is a crucial test in the evaluation of anemia. It helps distinguish between inadequate production and increased destruction or loss of RBCs:
- Low reticulocyte count: Suggests production problem (bone marrow failure, nutritional deficiency)
- High reticulocyte count: Suggests appropriate response to blood loss or hemolysis
Management and Nursing Care
Management of pediatric anemia is directed at the underlying cause:
| Type of Anemia | Management Strategies | Nursing Considerations |
|---|---|---|
| Iron Deficiency |
|
|
| Thalassemia |
|
|
| Sickle Cell Disease |
|
|
| B12/Folate Deficiency |
|
|
| Hemolytic Anemia |
|
|
Mnemonic: “IRON” – Key Interventions for Iron Deficiency Anemia
I Investigate cause (blood loss, poor intake, malabsorption)
R Replace iron (oral/parenteral supplementation)
O Optimize diet (iron-rich foods, vitamin C to enhance absorption)
N Note response (recheck hemoglobin after 4 weeks of therapy)
Warning Signs Requiring Immediate Attention
The following symptoms in a child with anemia may indicate the need for immediate medical intervention:
- Severe pallor with hemodynamic instability
- Respiratory distress
- Altered mental status
- Hemoglobin <6-7 g/dL with symptoms
- Active bleeding with hemodynamic compromise
- Acute splenic sequestration in sickle cell disease
Prevention Strategies
Iron Deficiency Prevention
- Exclusive breastfeeding for first 6 months
- Iron-fortified formula for non-breastfed infants
- Introduction of iron-rich complementary foods at 6 months
- Iron supplementation for premature infants
- Delay introduction of cow’s milk until 12 months
- Limit cow’s milk to 24 oz/day in toddlers
- Universal screening at 9-12 months and risk-based screening at other ages
Other Preventive Measures
- Genetic counseling: For families with hemoglobinopathies
- Newborn screening: For hemoglobinopathies like sickle cell disease
- Nutritional counseling: For balanced diet with adequate micronutrients
- Infection prevention: For children with sickle cell disease or thalassemia
- Vitamin supplementation: For high-risk groups (e.g., strict vegetarians for B12)
Nursing Roles in Anemia Management
- Screening and assessment: Recognize risk factors and early signs of anemia
- Education: Teach families about nutrition, medication administration, and disease management
- Monitoring: Follow growth parameters, medication adherence, and treatment response
- Support: Provide psychosocial support for children with chronic anemia
- Advocacy: Ensure access to needed services and resources
Pediatric Leukemia
Overview
Leukemia is the most common childhood cancer, accounting for approximately 30% of all pediatric malignancies. It is characterized by the uncontrolled proliferation of immature white blood cells (blasts) in the bone marrow, which interferes with normal hematopoiesis and may infiltrate extramedullary sites.
Clinical Pearl
Despite being a serious diagnosis, pediatric leukemia, particularly acute lymphoblastic leukemia (ALL), has one of the highest cure rates among childhood cancers, with more than 85% of children achieving long-term survival with current treatment protocols.
Classification of Pediatric Leukemia
Leukemia is classified based on the cell lineage affected and the degree of cellular maturation:

Figure 3: Classification and characteristics of pediatric leukemias
Acute Lymphoblastic Leukemia (ALL)
- Incidence: ~75-80% of childhood leukemias
- Peak age: 2-5 years
- Cell origin: Lymphoid precursors (B or T cell)
- Subtypes:
- B-cell ALL (80-85%)
- T-cell ALL (15-20%)
- Prognosis factors: Age, WBC count at diagnosis, cytogenetics, response to treatment
Acute Myeloid Leukemia (AML)
- Incidence: ~15-20% of childhood leukemias
- Age distribution: More uniform across childhood
- Cell origin: Myeloid precursors
- Subtypes: Based on FAB classification (M0-M7) or WHO classification
- Prognosis factors: Cytogenetics, molecular markers, response to induction chemotherapy
Other Types of Pediatric Leukemia
Chronic Myeloid Leukemia (CML)
- Rare in children (<5% of pediatric leukemias)
- Characterized by Philadelphia chromosome t(9;22)
- Three phases: chronic, accelerated, blast crisis
- Treatment with tyrosine kinase inhibitors
Juvenile Myelomonocytic Leukemia (JMML)
- Rare disorder of early childhood
- Features of both myelodysplastic and myeloproliferative disorders
- Associated with NF1, Noonan syndrome
- Treatment typically requires HSCT
Mnemonic: “ALL FACTS” – Risk Factors for Better Prognosis in ALL
A Age: 1-10 years
L Low initial WBC (<50,000/µL)
L Lack of CNS/testicular involvement
F Female gender
A Ancestry: Caucasian
C Chromosomal abnormalities (favorable ones like hyperdiploidy)
T Treatment response (rapid early response)
S Subtle or no organomegaly
Pathophysiology
Leukemia develops through a multistep process involving genetic alterations that lead to transformation of normal hematopoietic progenitors:
Key Pathophysiological Processes
- Genetic Alterations: Chromosomal translocations, deletions, duplications, and point mutations affecting oncogenes and tumor suppressors
- Clonal Proliferation: Uncontrolled proliferation of immature cells (blasts) that fail to differentiate properly
- Bone Marrow Failure: Displacement of normal hematopoietic cells resulting in cytopenias (anemia, thrombocytopenia, neutropenia)
- Extramedullary Spread: Infiltration of leukemic cells into organs and tissues outside the bone marrow
Common Genetic Alterations in ALL
- Favorable:
- Hyperdiploidy (>50 chromosomes)
- t(12;21) ETV6-RUNX1 fusion
- Unfavorable:
- Hypodiploidy (<44 chromosomes)
- t(9;22) BCR-ABL1 fusion (Philadelphia chromosome)
- KMT2A (MLL) rearrangements
- iAMP21 (intrachromosomal amplification of chromosome 21)
Common Genetic Alterations in AML
- Favorable:
- t(8;21) RUNX1-RUNX1T1 fusion
- inv(16) CBFB-MYH11 fusion
- t(15;17) PML-RARA fusion (Acute Promyelocytic Leukemia)
- NPM1 mutation without FLT3-ITD
- Unfavorable:
- Complex karyotype
- Monosomy 7
- FLT3-ITD mutation
- KMT2A (MLL) rearrangements
Important Note
Genetic and molecular testing are critical for risk stratification and treatment decisions in pediatric leukemia. Modern treatment protocols incorporate cytogenetic and molecular findings to determine therapy intensity.
Clinical Manifestations
The clinical manifestations of pediatric leukemia result from bone marrow failure, tumor burden, and organ infiltration:
Bone Marrow Failure
- Anemia: Pallor, fatigue, weakness, tachycardia
- Thrombocytopenia: Petechiae, bruising, bleeding (gums, nose)
- Neutropenia: Recurrent or persistent infections, fever
Tumor Burden
- Hepatosplenomegaly: Abdominal distension, discomfort
- Lymphadenopathy: Painless, firm enlargement of lymph nodes
- Bone pain: Due to marrow expansion or periosteal infiltration
Organ Infiltration
- CNS: Headache, vomiting, visual changes, cranial nerve palsies
- Mediastinum: Cough, dyspnea, superior vena cava syndrome (in T-cell ALL)
- Testes: Painless testicular enlargement
Constitutional Symptoms
- Fever (may be persistent or intermittent)
- Weight loss
- Night sweats
- Malaise
- Anorexia
- Irritability
Clinical Pearl
Bone pain in children with leukemia may be misdiagnosed as “growing pains” or rheumatic disorders. Consider leukemia in a child with persistent or severe bone pain, especially when accompanied by other signs like pallor, bruising, or persistent infections.
Diagnostic Approach
The diagnostic evaluation of a child with suspected leukemia includes:
| Diagnostic Test | Purpose | Findings in Leukemia |
|---|---|---|
| Complete Blood Count (CBC) | Initial screening test | Anemia, thrombocytopenia, abnormal WBC (high, normal, or low with blasts) |
| Peripheral Blood Smear | Evaluate cell morphology | Presence of blasts, abnormal cell morphology |
| Bone Marrow Aspiration and Biopsy | Definitive diagnosis | >20% blasts in bone marrow confirms leukemia |
| Immunophenotyping (Flow Cytometry) | Determine cell lineage and subtype | B-cell vs. T-cell ALL; AML subtypes; expression of specific markers |
| Cytogenetic Analysis | Detect chromosomal abnormalities | Translocations, deletions, additions; ploidy status |
| Molecular Studies | Identify specific genetic alterations | Gene fusions, mutations (FLT3, NPM1, etc.) |
| Lumbar Puncture | Assess CNS involvement | Presence of blasts in CSF; CSF protein and glucose |
| Imaging Studies | Evaluate disease extent | Hepatosplenomegaly, lymphadenopathy, mediastinal mass |
Mnemonic: “LEUKEMIA” – Key Diagnostic Steps
L Look at CBC and peripheral smear
E Examine bone marrow (aspiration and biopsy)
U Understand cell lineage (immunophenotyping)
K Karyotype (cytogenetic analysis)
E Evaluate molecular markers
M Measure CNS involvement (lumbar puncture)
I Imaging for disease extent
A Assess for organ dysfunction
Treatment Approach
Treatment of pediatric leukemia involves a multidisciplinary approach and typically includes:
Treatment Phases for ALL
- Remission Induction (4-6 weeks): Achieve complete remission (CR) with intensive chemotherapy
- Consolidation/Intensification (2-8 months): Eradicate residual leukemic cells
- CNS-directed Therapy: Prevent CNS relapse with intrathecal chemotherapy and/or radiation
- Maintenance (2-3 years): Prevent relapse with less intensive, prolonged therapy
Treatment Approach for AML
- Induction (1-2 cycles): Achieve remission with intensive chemotherapy
- Consolidation (multiple cycles): Intensive chemotherapy to eliminate residual disease
- CNS Prophylaxis: Intrathecal chemotherapy
- Hematopoietic Stem Cell Transplantation (HSCT): For high-risk patients or after relapse
Common Chemotherapy Agents
- ALL:
- Vincristine
- Prednisone/Dexamethasone
- L-Asparaginase
- Doxorubicin
- Methotrexate
- 6-Mercaptopurine
- Cyclophosphamide
- AML:
- Cytarabine (high-dose)
- Daunorubicin/Idarubicin
- Etoposide
- Mitoxantrone
Targeted Therapies
- Tyrosine Kinase Inhibitors: For Philadelphia chromosome-positive ALL (imatinib, dasatinib)
- FLT3 Inhibitors: For FLT3-mutated AML (midostaurin, gilteritinib)
- Monoclonal Antibodies: (blinatumomab, inotuzumab)
- CAR-T Cell Therapy: For refractory/relapsed B-cell ALL
- All-trans Retinoic Acid (ATRA): For acute promyelocytic leukemia
Clinical Pearl
Risk-adapted therapy is a cornerstone of modern pediatric leukemia treatment. Children are stratified into risk groups (standard, intermediate, high, or very high) based on clinical, biological, and response factors, with treatment intensity tailored accordingly.
Nursing Management
Nursing care for children with leukemia is complex and addresses multiple dimensions of care:
Supportive Care
- Infection Prevention:
- Neutropenic precautions
- Hand hygiene
- Isolation as needed
- Monitoring for signs of infection
- Bleeding Precautions:
- Platelet transfusions as needed
- Safety measures to prevent injury
- Monitoring for bleeding signs
- Nutritional Support:
- Dietary consultation
- Enteral or parenteral nutrition
- Monitoring growth parameters
Symptom Management
- Pain Management:
- Assessment using age-appropriate scales
- Pharmacological interventions
- Non-pharmacological techniques
- Nausea and Vomiting:
- Antiemetics (pre-medication)
- Hydration
- Environmental modifications
- Mucositis:
- Oral hygiene
- Pain relief (topical, systemic)
- Nutritional adaptations
Psychosocial Support
- Child:
- Age-appropriate education
- Play therapy
- Coping strategies
- School reintegration
- Family:
- Education about disease and treatment
- Emotional support
- Resource referrals
- Sibling support
- Resources:
- Financial assistance
- Support groups
- Camp programs
Managing Treatment Complications
Acute Complications
- Tumor Lysis Syndrome: Hydration, allopurinol/rasburicase, electrolyte monitoring
- Neutropenic Fever: Prompt antibiotic therapy, culture collection
- Hemorrhage: Blood product administration, pressure, hemostatic agents
- Anaphylaxis: (e.g., to L-asparaginase) Emergency management, preparation for future doses
Long-term Complications
- Cardiotoxicity: Monitoring, cardiac protective strategies
- Neurocognitive Effects: Neuropsychological assessment, educational support
- Growth Impairment: Growth monitoring, endocrine referral
- Infertility: Age-appropriate discussion, fertility preservation options
- Secondary Malignancies: Surveillance, education about warning signs
Mnemonic: “PROTECT” – Nursing Priorities in Leukemia Care
P Prevent infection (neutropenia precautions)
R Recognize complications early
O Observe for bleeding (thrombocytopenia)
T Treat symptoms (pain, nausea, mucositis)
E Educate patient and family
C Care coordination across settings
T Tailor psychosocial support
Long-term Follow-up and Survivorship
As survival rates for pediatric leukemia continue to improve, long-term follow-up care becomes increasingly important:
Surveillance for Late Effects
- Cardiac: Echocardiograms, ECGs (anthracycline exposure)
- Endocrine: Growth, thyroid, gonadal function
- Neurocognitive: Educational assessment, neuropsychological testing
- Renal/Hepatic: Function tests
- Secondary Malignancies: Risk-based screening
- Psychosocial: Depression, anxiety, post-traumatic stress
Survivorship Care
- Individualized Survivorship Plan: Summarizing treatment and follow-up recommendations
- Health Promotion: Healthy lifestyle, risk reduction
- Education: About potential late effects and warning signs
- Transition: To adult-focused care
- Advocacy: Educational, employment, insurance issues
- Support Services: Survivor networks, counseling
Clinical Pearl: The Success Story of Pediatric ALL
The transformation of childhood ALL from a virtually incurable disease in the 1950s to one with >85% cure rates today represents one of the greatest success stories in modern medicine. This success has been achieved through:
- Systematic clinical trials
- Risk-stratified therapy
- Effective CNS prophylaxis
- Improved supportive care
- Molecular characterization leading to targeted therapies